Defiant Quick-Start Guide
Warning
The Defiant documentation is being actively updated. If you have anything you would like to add please reach out to help@olcf.ornl.gov.
System Overview
“Defiant2” is the all new hardware refresh of the original “Defiant” system; however, the name of the cluster will remain Defiant. Defiant is an NCCS open access system and is one of the Advanced Computing Ecosystem (ACE) testbeds that provides a sandbox for testing advances in IRI workflows and patterns, emerging compute, network, and storage architectures, and cloudification of traditional HPC use-cases. The system currently consists of 20 CPU, 2 GPU nodes and 1 login node for user code compilations.
Defiant Nodes
The system consists of 20 HPE Cray XD220 CPU nodes along with 2 HPE Cray XD670 Nvidia H200 GPU nodes. It also has 1 login node with an Nvidia H100 for user code compilations.
GPU Compute Nodes
Each Defiant GPU compute node consists of [2x] 32-core INTEL(R) XEON(R) PLATINUM 8562Y+ CPUs with 2 hardware threads per physical core. Both CPUs have access to 4TB of DDR5 memory and [8x] 144GB NVIDIA H200 GPUs. The GPU nodes have [8x] 400g Infiniband interconnects. Additionally, there are [2x] 100g connections for Data Streaming. Each node also contains [4x] 3.5TB NVME devices (SSDs).
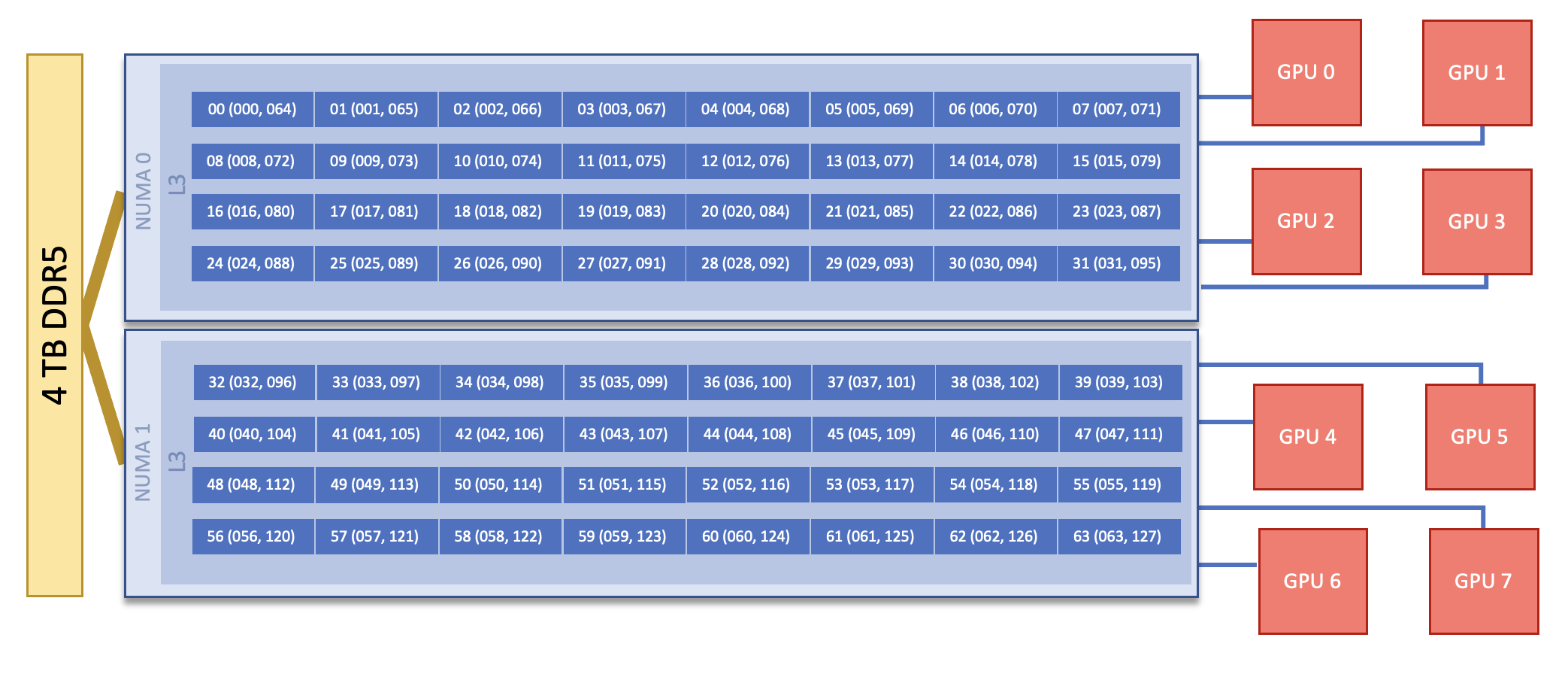
Note
There are 2 NUMA domains per node, that are defined as follows:
NUMA 0: hardware threads 0-31, 64-95
NUMA 1: hardware threads 32-63, 96-127
CPU Compute Nodes
Each Defiant CPU compute node consists of [2x] 32-core INTEL(R) XEON(R) PLATINUM 8462Y+ CPUs with 2 hardware threads per physical core. Both CPUs have access to 512GB of DDR5 memory. The nodes have [1x] 400g Infiniband interconnect per node. Additionally, there are [1x] 25g connections for Data Streaming.
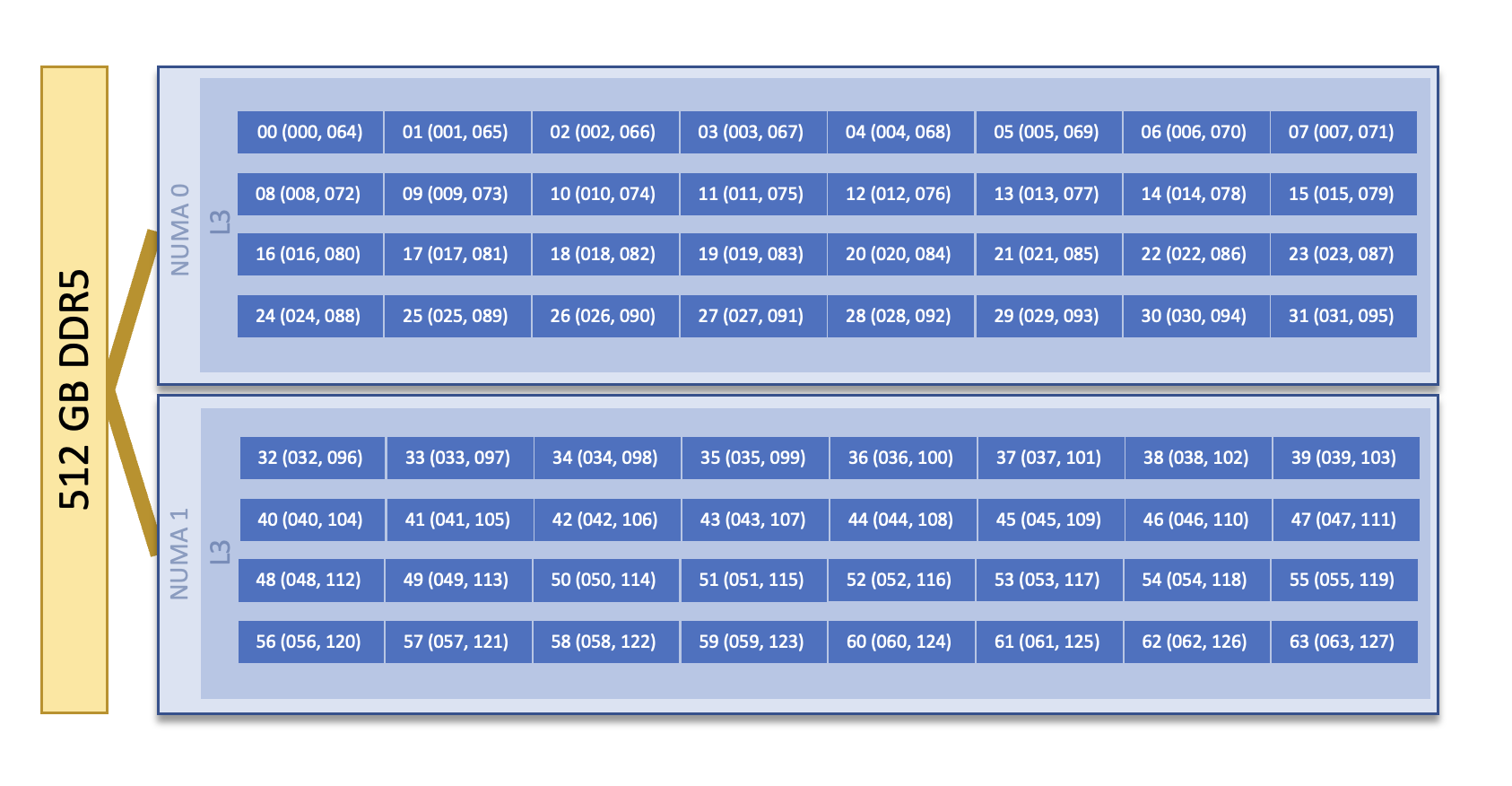
Note
There are 2 NUMA domains per node, that are defined as follows:
NUMA 0: hardware threads 0-31, 64-95
NUMA 1: hardware threads 32-63, 96-127
Login Node
The Defiant login node consists of [2x] 32-core INTEL(R) XEON(R) PLATINUM 8462Y+ CPUs with 2 hardware threads per physical core. Both CPUs have access to 384GB of DDR5 memory and [1x] 80GB NVIDIA H100 GPU. The login node has [1x] 400g Infiniband interconnect. Additionally, there are [2x] 100g connections for Data Streaming. The login node also contains a 890GB NVME device (SSD).
System Interconnect
The Defiant nodes are connected with 400g NDR Mellanox ConnectX-7 NIC Infiniband interconnects in a Leaf & Spine configuration.
File Systems
Defiant is connected to the Lustre Polis filesystem providing ~3.6 PB of usable namespace (/lustre/polis/).
Defiant also has access to the center-wide NFS-based filesystem (which provides user and project home areas) in /ccsopen/home/<username>
GPUs
Defiant contains a total of 16 NVIDIA H200 GPUs. Based on the Hopper Architecture, the NVIDIA H200 GPU has a peak performance of up to 4 PFLOPS in FP8. Each GPU contains 144 GB HBM3e Memory which can be accessed at speeds of 4.8 TB/s.
Connecting
To connect to Defiant, first ssh to the jump server: flux.op.ccs.ornl.gov and then to the login node: login1.defiant.ccs.ornl.gov. For example:
$ ssh username@flux.op.ccs.ornl.gov
$ ssh username@login1.defiant.ccs.ornl.gov
Data and Storage
For more detailed information about center-wide file systems and data archiving available on Defiant, please refer to the pages on Data Storage and Transfers, but the two subsections below give a quick overview of NFS and GPFS storage spaces.
NFS Filesystem
Area |
Path |
Type |
Permissions |
Quota |
Backups |
Purge |
Retention |
On Compute Nodes |
|---|---|---|---|---|---|---|---|---|
User Home |
|
NFS |
User set |
50 GB |
Yes |
No |
90 days |
yes |
Note
Please not that this /ccsopen location is not the same NFS filesystem as found in other Open enclave systems like Odo. So files
created on Defiant will not be available on Odo.
Lustre Filesystem (Polis)
Area |
Path |
Type |
Permissions |
Quota |
Backups |
Purge |
Retention |
On Compute Nodes |
|---|---|---|---|---|---|---|---|---|
Member Work |
|
Lustre HPE ClusterStor |
700 |
50 TB |
No |
90 days |
N/A |
yes |
Project Work |
|
Lustre HPE ClusterStor |
770 |
50 TB |
No |
90 days |
N/A |
yes |
World Work |
|
Lustre HPE ClusterStor |
770 |
50 TB |
No |
90 days |
N/A |
yes |
Programming Environment
OLCF provides Defiant users many pre-installed software packages and scientific libraries. To facilitate this, environment management tools are used to handle necessary changes to the shell.
Environment Modules (Lmod)
Environment modules are provided through Lmod, a Lua-based module system for
dynamically altering shell environments. By managing changes to the shell’s
environment variables (such as PATH, LD_LIBRARY_PATH, and
PKG_CONFIG_PATH), Lmod allows you to alter the software available in your
shell environment without the risk of creating package and version combinations
that cannot coexist in a single environment.
General Usage
The interface to Lmod is provided by the module command:
Command |
Description |
|---|---|
|
Shows a terse list of the currently loaded modules |
|
Shows a table of the currently available modules |
|
Shows help information about |
|
Shows the environment changes made by the |
|
Searches all possible modules according to |
|
Loads the given |
|
Adds |
|
Removes |
|
Unloads all modules |
|
Resets loaded modules to system defaults |
|
Reloads all currently loaded modules |
Searching for Modules
Modules with dependencies are only available when the underlying dependencies,
such as compiler families, are loaded. Thus, module avail will only display
modules that are compatible with the current state of the environment. To
search the entire hierarchy across all possible dependencies, the spider
sub-command can be used as summarized in the following table.
Command |
Description |
|---|---|
|
Shows the entire possible graph of modules |
|
Searches for modules named |
|
Searches for a specific version of |
|
Searches for modulefiles containing |
Compilers
Cray, NVIDIA, Intel, and GCC compilers are provided through modules on Defiant. The system GCC (version 11.5.0) compiler is also located in
/usr/bin. The table below lists details about each of the module-provided compilers.
Note
It is highly recommended to use the Cray compiler wrappers (cc, CC, and ftn) whenever possible. See the next section for more details.
Vendor |
Programming Environment |
Compiler Module |
Language |
Compiler Wrapper |
Compiler |
|---|---|---|---|---|---|
Cray |
|
|
C |
|
|
C++ |
|
|
|||
Fortran |
|
|
|||
NVIDIA |
|
|
C |
|
|
C++ |
|
|
|||
Fortran |
|
|
|||
Intel |
|
|
C |
|
|
C++ |
|
|
|||
Fortran |
|
|
|||
GCC |
|
|
C |
|
|
C++ |
|
|
|||
Fortran |
|
|
Cray Programming Environment and Compiler Wrappers
Cray provides PrgEnv-<compiler> modules (e.g., PrgEnv-cray) that load
compatible components of a specific compiler toolchain. The components include
the specified compiler as well as MPI, LibSci, and other libraries. Loading the
PrgEnv-<compiler> modules also defines a set of compiler wrappers for that
compiler toolchain that automatically add include paths and link in libraries
for Cray software. Compiler wrappers are provided for C (cc), C++ (CC),
and Fortran (ftn).
Note
Use the -craype-verbose flag to display the full include and link information
used by the Cray compiler wrappers. This must be called on a file to see the full
output (e.g., CC -craype-verbose test.cpp).
MPI
The MPI implementation available on Defiant is Cray’s MPICH, which is “GPU-aware” so GPU buffers can be passed directly to MPI calls. Currently, Defiant has MPICH versions 8.1.32 as default and 9.0.0 as an additional module.
Starting from the 8.1.26 release, Cray MPICH supports the Intel Sapphire Rapids CPU HBM processor architecture
Compiling
This section covers how to compile for different programming models using the different compilers covered in the previous section.
MPI
Implementation |
Module |
Compiler |
Header Files & Linking |
|---|---|---|---|
Cray MPICH |
|
|
MPI header files and linking is built into the Cray compiler wrappers |
|
-I$(MPICH_DIR)/include-L$(MPICH_DIR)/lib -lmpi |
GPU-Aware MPI
1. Compiling with the Cray compiler wrappers, cc or CC
To use GPU-aware Cray MPICH with the Cray compiler wrappers, users must load specific modules, set some environment variables, and include appropriate headers and libraries. The following modules and environment variables must be set:
#Load your Programming Environment of Choice
module load [PrgEnv-cray, PrgEnv-nvidia, PrgEnv-gnu, PrgEnv-intel]
module load cuda/12.6
module load craype-accel-nvidia90
## These must be set before running
export MPICH_GPU_SUPPORT_ENABLED=1
In addition, the HPE Cray MPI compiler wrappers are not aware of the HPE Cray MPI GTL libraries for use with GPU codes, they can be simply used to compile for use with GPUs. Relative symlinks are provided in the library directories pointing to the location of the gtl libraries within a given version of HPE Cray MPI. Simply link the appropriate gtl library for your intended GPU.
-lmpi_gtl_cuda
You will also likely need #include <cuda_runtime_api.h> included in the source file.
OpenMP
This section shows how to compile with OpenMP using the different compilers covered above.
Vendor |
Module |
Language |
Compiler |
OpenMP flag (CPU thread) |
|---|---|---|---|---|
Cray |
|
C, C++ |
ccCC |
|
Fortran |
|
-homp-fopenmp (alias) |
||
NVIDIA |
|
C
C++
Fortran
|
nvcnvc++nvfortan |
|
Intel |
|
C
C++
Fortran
|
icxicpxifx |
|
GCC |
|
C
C++
Fortran
|
$GCC_PATH/bin/gcc$GCC_PATH/bin/g++$GCC_PATH/bin/gfortran |
|
OpenMP GPU Offload
This section shows how to compile with OpenMP Offload using the different compilers covered above.
Note
Make sure the craype-accel-nvidia90 & cuda modules are loaded when using OpenMP offload.
Vendor |
Module |
Language |
Compiler |
OpenMP flag (GPU) |
|---|---|---|---|---|
Cray |
|
C C++ |
ccCC |
|
Fortran |
|
-homp-fopenmp (alias) |
||
NVIDIA |
|
C
C++
Fortran
|
nvcnvc++nvfortan |
|
Intel |
|
C
C++
Fortran
|
icxicpxifx |
|
Running Jobs
This section describes how to run programs on the Defiant compute nodes, including a brief overview of Slurm and also how to map processes and threads to CPU cores and GPUs.
Slurm Workload Manager
Slurm is the workload manager used to interact with the compute nodes on Defiant. In the following subsections, the most commonly used Slurm commands for submitting, running, and monitoring jobs will be covered, but users are encouraged to visit the official documentation and man pages for more information.
Batch Scheduler and Job Launcher
Slurm provides 3 ways of submitting and launching jobs on Defiant’s compute nodes: batch scripts, interactive, and single-command. The Slurm commands associated with these methods are shown in the table below and examples of their use can be found in the related subsections.
|
Used to submit a batch script to allocate a Slurm job allocation. The script contains options preceded with
#SBATCH.(see Batch Scripts section below)
|
|
Used to allocate an interactive Slurm job allocation, where one or more job steps (i.e.,
srun commands) can then be launched on the allocated resources (i.e., nodes).(see Interactive Jobs section below)
|
|
Used to run a parallel job (job step) on the resources allocated with sbatch or
salloc.If necessary, srun will first create a resource allocation in which to run the parallel job(s).
(see Single Command section below)
|
Batch Scripts
A batch script can be used to submit a job to run on the compute nodes at a later time. In this case, stdout and stderr will be written to a file(s) that can be opened after the job completes. Here is an example of a simple batch script:
1#!/bin/bash
2#SBATCH -A <project_id>
3#SBATCH -J <job_name>
4#SBATCH -o %x-%j.out
5#SBATCH -t 00:05:00
6#SBATCH -p <partition>
7#SBATCH -N 2
8
9srun -n4 --ntasks-per-node=2 ./a.out
The Slurm submission options are preceded by #SBATCH, making them appear as
comments to a shell (since comments begin with #). Slurm will look for
submission options from the first line through the first non-comment line.
Options encountered after the first non-comment line will not be read by Slurm.
In the example script, the lines are:
Line |
Description |
|---|---|
1 |
[Optional] shell interpreter line |
2 |
OLCF project to charge |
3 |
Job name |
4 |
stdout file name ( |
5 |
Walltime requested ( |
6 |
Batch queue |
7 |
Number of compute nodes requested |
8 |
Blank line |
9 |
|
Interactive Jobs
To request an interactive job where multiple job steps (i.e., multiple srun
commands) can be launched on the allocated compute node(s), the salloc
command can be used:
$ salloc -A <project_id> -J <job_name> -t 00:05:00 -p <partition> -N 2
salloc: Granted job allocation 4258
salloc: Waiting for resource configuration
salloc: Nodes defiant[10-11] are ready for job
$ srun -n 4 --ntasks-per-node=2 ./a.out
<output printed to terminal>
$ srun -n 2 --ntasks-per-node=1 ./a.out
<output printed to terminal>
Here, salloc is used to request an allocation of compute nodes for
5 minutes. Once the resources become available, the user is granted access to
the compute nodes (defiant10 and defiant11 in this case) and can launch job
steps on them using srun.
Single Command (non-interactive)
$ srun -A <project_id> -t 00:05:00 -p <partition> -N 2 -n 4 --ntasks-per-node=2 ./a.out
<output printed to terminal>
The job name and output options have been removed since stdout/stderr are typically desired in the terminal window in this usage mode.
Common Slurm Submission Options
The table below summarizes commonly-used Slurm job submission options:
|
Project ID to charge |
|
Name of job |
|
Partition / batch queue |
|
Wall clock time < |
|
Number of compute nodes |
|
Standard output file name |
|
Standard error file name |
For more information about these and/or other options, please see the
sbatch man page.
Other Common Slurm Commands
The table below summarizes commonly-used Slurm commands:
|
Used to view partition and node information.
E.g., to view user-defined details about the caar queue:
sinfo -p caar -o "%15N %10D %10P %10a %10c %10z" |
|
Used to view job and job step information for jobs in the scheduling queue.
E.g., to see all jobs from a specific user:
squeue -l -u <user_id> |
|
Used to view accounting data for jobs and job steps in the job accounting log (currently in the queue or recently completed).
E.g., to see a list of specified information about all jobs submitted/run by a users since 1 PM on January 4, 2021:
sacct -u <username> -S 2021-01-04T13:00:00 -o "jobid%5,jobname%25,user%15,nodelist%20" -X |
|
Used to signal or cancel jobs or job steps.
E.g., to cancel a job:
scancel <jobid> |
|
Used to view or modify job configuration.
E.g., to place a job on hold:
scontrol hold <jobid> |
Slurm Compute Node Partitions
Defiant’s compute nodes are separated into 2 Slurm partitions (queues): 1 for CPU jobs and 1 for GPU. Please see the tables below for details.
PARTITION |
NODELIST |
|---|---|
batch-cpu |
defiant[01-20] |
batch-gpu |
defiant-nv[01-02] |
GPU Partition
Defiant’s GPU partition consists of 2 nodes with 8 NVIDIA H200 GPUs each and
these nodes can be shared by multiple users, provided that the resources are
available. Therefore, users are required to specify the number of GPUs needed
for their job using the --gpus=<int> or --gres=gpu:<int> Slurm
submission options when submitting to the GPU partition. For example, the
submit script below requests 4 GPUs and launches 4 MPI ranks with 1 GPU each
1#!/bin/bash
2#SBATCH -A <project_id>
3#SBATCH -J <job_name>
4#SBATCH -o %x-%j.out
5#SBATCH -t 00:05:00
6#SBATCH -p batch-gpu
7#SBATCH -N 1
8#SBATCH --gpus=4
9
10srun -n 4 --gpus-per-task=1 --gpu-bind=closest ./a.out
Specifically, the GPU partition is requested by #SBATCH -p batch-gpu and
the number of GPUs is specified by #SBATCH --gpus=4. The srun flag
--gpus-per-task=1 specifies that each MPI rank should be allocated 1 GPU,
and the --gpu-bind=closest flag is used to bind each MPI rank to the
closest GPU available to that rank. Please refer to the
node diagram for more information about GPU locations
on the node. More information on the GPU-related srun flags can be found
in the GPU Mapping section.
Process and Thread Mapping
This section describes how to map processes (e.g., MPI ranks) and process threads (e.g., OpenMP threads) to the CPUs and GPUs on Defiant. The Defiant Nodes diagram will be helpful when reading this section to understand which hardware threads your processes and threads run on.
CPU Mapping
In this sub-section, a simple MPI+OpenMP “Hello, World” program
(hello_mpi_omp) will be used to
clarify the mappings. Slurm’s Interactive Jobs method was used to request an
allocation of 1 compute node for these examples:
salloc -A <project_id> -t 30 -p <parition> -N 1
The srun options used in this section are (see man srun for more
information):
|
Request that
ncpus be allocated per process (default is 1).(
ncpus refers to hardware threads) |
|
In task layout, use the specified maximum number of threads per core
(default is 1; there are 2 hardware threads per physical CPU core).
|
|
Bind tasks to CPUs.
threads - Automatically generate masks binding tasks to threads.(Although this option is not explicitly used in these examples, it is the default CPU binding.)
|
Note
In the srun man page (and so the table above), threads refers
to hardware threads.
2 MPI ranks - each with 2 OpenMP threads
In this example, the intent is to launch 2 MPI ranks, each of which spawn 2 OpenMP threads, and have all of the 4 OpenMP threads run on different physical CPU cores.
First (INCORRECT) attempt
To set the number of OpenMP threads spawned per MPI rank, the
OMP_NUM_THREADS environment variable can be used. To set the number
of MPI ranks launched, the srun flag -n can be used.
$ export OMP_NUM_THREADS=2
$ srun -n2 ./hello_mpi_omp | sort
WARNING: Requested total thread count and/or thread affinity may result in
oversubscription of available CPU resources! Performance may be degraded.
Explicitly set OMP_WAIT_POLICY=PASSIVE or ACTIVE to suppress this message.
Set CRAY_OMP_CHECK_AFFINITY=TRUE to print detailed thread-affinity messages.
WARNING: Requested total thread count and/or thread affinity may result in
oversubscription of available CPU resources! Performance may be degraded.
Explicitly set OMP_WAIT_POLICY=PASSIVE or ACTIVE to suppress this message.
Set CRAY_OMP_CHECK_AFFINITY=TRUE to print detailed thread-affinity messages.
MPI 000 - OMP 000 - HWT 000 - Node defiant01
MPI 000 - OMP 001 - HWT 000 - Node defiant01
MPI 001 - OMP 000 - HWT 016 - Node defiant01
MPI 001 - OMP 001 - HWT 016 - Node defiant01
The first thing to notice here is the WARNING about oversubscribing the
available CPU cores. Also, the output shows each MPI rank did spawn 2 OpenMP
threads, but both OpenMP threads ran on the same hardware thread (for a given
MPI rank). This was not the intended behavior; each OpenMP thread was meant
to run on its own physical CPU core.
Second (CORRECT) attempt
By default, each MPI rank is allocated only 1 hardware thread, so both OpenMP
threads only have that 1 hardware thread to run on - hence the WARNING and
undesired behavior. In order for each OpenMP thread to run on its own physical
CPU core, each MPI rank should be given 2 hardware thread (-c 2) - since,
by default, only 1 hardware thread per physical CPU core is enabled (this would
need to be -c 4 if --threads-per-core=2 instead of the default of 1.
The OpenMP threads will be mapped to unique physical CPU cores unless there are
not enough physical CPU cores available, in which case the remaining OpenMP
threads will share hardware threads and a WARNING will be issued as shown in
the previous example.
$ export OMP_NUM_THREADS=2
$ srun -n2 -c2 ./hello_mpi_omp | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant11
MPI 000 - OMP 001 - HWT 001 - Node defiant11
MPI 001 - OMP 000 - HWT 032 - Node defiant11
MPI 001 - OMP 001 - HWT 033 - Node defiant11
Now the output shows that each OpenMP thread ran on (one of the hardware threads of) its own physical CPU cores. More specifically (see the Defiant Compute Node diagram), OpenMP thread 000 of MPI rank 000 ran on hardware thread 000 (i.e., physical CPU core 00), OpenMP thread 001 of MPI rank 000 ran on hardware thread 001 (i.e., physical CPU core 01), OpenMP thread 000 of MPI rank 001 ran on hardware thread 032 (i.e., physical CPU core 32), and OpenMP thread 001 of MPI rank 001 ran on hardware thread 033 (i.e., physical CPU core 33) - as expected.
Note
There are many different ways users might choose to perform these mappings,
so users are encouraged to clone the hello_mpi_omp program and test
whether or not processes and threads are running where intended.
GPU Mapping
In this sub-section, an MPI+OpenMP+CUDA “Hello, World” program, hello_jobstep.cpp, will be used to clarify the GPU mappings.
1 /**********************************************************
2 "Hello World"-type program to test different srun layouts.
3
4 Written by Tom Papatheodore
5 **********************************************************/
6
7 #include <stdlib.h>
8 #include <stdio.h>
9 #include <iostream>
10 #include <iomanip>
11 #include <iomanip>
12 #include <string.h>
13 #include <mpi.h>
14 #include <sched.h>
15 #include <cuda.h>
16 #include <cuda_runtime_api.h>
17 #include <omp.h>
18
19 // Macro for checking errors in HIP API calls
20 #define cudaErrorCheck(call) \
21 do{ \
22 cudaError_t cudaErr = call; \
23 if(cudaSuccess != cudaErr){ \
24 printf("CUDA Error - %s:%d: '%s'\n", __FILE__, __LINE__, cudaGetErrorString(cudaErr)); \
25 exit(0); \
26 } \
27 }while(0)
28
29 int main(int argc, char *argv[]){
30
31 MPI_Init(&argc, &argv);
32
33 int size;
34 MPI_Comm_size(MPI_COMM_WORLD, &size);
35
36 int rank;
37 MPI_Comm_rank(MPI_COMM_WORLD, &rank);
38
39 char name[MPI_MAX_PROCESSOR_NAME];
40 int resultlength;
41 MPI_Get_processor_name(name, &resultlength);
42
43 // If CUDA_VISIBLE_DEVICES is set, capture visible GPUs
44 const char* gpu_id_list;
45 const char* cuda_visible_devices = getenv("CUDA_VISIBLE_DEVICES");
46 if(cuda_visible_devices == NULL){
47 gpu_id_list = "N/A";
48 }
49 else{
50 gpu_id_list = cuda_visible_devices;
51 }
52
53 // Find how many GPUs HIP runtime says are available
54 int num_devices = 0;
55 cudaErrorCheck( cudaGetDeviceCount(&num_devices) );
56
57 int hwthread;
58 int thread_id = 0;
59
60 if(num_devices == 0){
61 #pragma omp parallel default(shared) private(hwthread, thread_id)
62 {
63 thread_id = omp_get_thread_num();
64 hwthread = sched_getcpu();
65
66 printf("MPI %03d - OMP %03d - HWT %03d - Node %s\n",
67 rank, thread_id, hwthread, name);
68
69 }
70 }
71 else{
72
73 char busid[64];
74
75 std::string busid_list = "";
76 std::string rt_gpu_id_list = "";
77
78 // Loop over the GPUs available to each MPI rank
79 for(int i=0; i<num_devices; i++){
80
81 cudaErrorCheck( cudaSetDevice(i) );
82
83 // Get the PCIBusId for each GPU and use it to query for UUID
84 cudaErrorCheck( cudaDeviceGetPCIBusId(busid, 64, i) );
85
86 // Concatenate per-MPIrank GPU info into strings for print
87 if(i > 0) rt_gpu_id_list.append(",");
88 rt_gpu_id_list.append(std::to_string(i));
89
90 std::string temp_busid(busid);
91
92 if(i > 0) busid_list.append(",");
93 busid_list.append(temp_busid.substr(5,2));
94
95 }
96
97 #pragma omp parallel default(shared) private(hwthread, thread_id)
98 {
99 #pragma omp critical
100 {
101 thread_id = omp_get_thread_num();
102 hwthread = sched_getcpu();
103
104 printf("MPI %03d - OMP %03d - HWT %03d - Node %s - RT_GPU_ID %s - GPU_ID %s - Bus_ID %s\n",
105 rank, thread_id, hwthread, name, rt_gpu_id_list.c_str(), gpu_id_list, busid_list.c_str());
106 }
107 }
108 }
109
110 MPI_Finalize();
111
112 return 0;
113 }
Makefile
1 COMP = CC
2
3 CFLAGS = -std=c++11 -fopenmp
4 LFLAGS = -fopenmp
5
6 INCLUDES = -I${MPICH_DIR}/include
7 LIBRARIES = -L${MPICH_DIR}/lib
8
9 hello_jobstep: hello_jobstep.o
10 ${COMP} ${LFLAGS} ${LIBRARIES} hello_jobstep.o -o hello_jobstep
11
12 hello_jobstep.o: hello_jobstep.cpp
13 ${COMP} ${CFLAGS} ${INCLUDES} -c hello_jobstep.cpp
14
15 .PHONY: clean
16
17 clean:
18 rm -f hello_jobstep *.o
Again, Slurm’s Interactive Jobs method was used to
request an allocation of 2 compute node for these examples:
salloc -A <project_id> -t 30 -p <parition> -N 2. The CPU mapping part of
this example is very similar to the example used above in the CPU Mapping
sub-section, so the focus here will be on the GPU mapping part.
The following srun options will be used in the examples below. See
man srun for a complete list of options and more information.
|
Specify the number of GPUs required for the job on each task to be spawned in the job’s resource allocation. |
|
Bind tasks to specific GPUs by setting GPU masks on tasks (or ranks) as specified where
|
|
Bind all GPUs to all tasks |
|
Request that there are ntasks tasks invoked for every GPU. |
|
Specifies the distribution of MPI ranks across compute nodes, sockets (NUMA domains on Defiant), and cores,
respectively. The default values are |
Note
In general, GPU mapping can be accomplished in different ways. For example, an
application might map MPI ranks to GPUs programmatically within the code using,
say, cudaSetDevice. In this case, since all GPUs on a node are available to
all MPI ranks on that node by default, there might not be a need to map to GPUs
using Slurm (just do it in the code). However, in another application, there
might be a reason to make only a subset of GPUs available to the MPI ranks on a
node. It is this latter case that the following examples refer to.
Mapping 1 task per GPU
In the following examples, each MPI rank (and its OpenMP threads) will be mapped to a single GPU.
Example 1: 4 MPI ranks - each with 2 OpenMP threads and 1 GPU (single-node)
This example launches 4 MPI ranks (-n4), each with 2 physical CPU cores
(-c2) to launch 2 OpenMP threads (OMP_NUM_THREADS=2) on. In addition,
each MPI rank (and its 2 OpenMP threads) should have access to only 1 GPU. To
accomplish the GPU mapping, one new srun options will be used:
--gpus-per-taskspecifies the number of GPUs required for the job on each task
Note
To further clarify, --gpus-per-task does not actually bind GPUs to MPI ranks.
It allocates GPUs to the job step. The default GPU map is what actually
maps a specific GPU to each rank.
(see the Defiant Nodes section).
$ export OMP_NUM_THREADS=2
$ srun -N1 -n4 -c2 --gpus-per-task=1 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 000 - OMP 001 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 002 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 001 - OMP 001 - HWT 003 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 002 - OMP 000 - HWT 004 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 002 - OMP 001 - HWT 005 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 003 - OMP 000 - HWT 006 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 003 - OMP 001 - HWT 007 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
The output contains different IDs associated with the GPUs so it is important to
first describe these IDs before moving on. GPU_ID is the node-level (or global)
GPU ID, which is labeled as one might expect from looking at a node diagram:
0, 1, 2, 3. RT_GPU_ID is the CUDA runtime GPU ID, which can be thought of as
each MPI rank’s local GPU ID numbering (with zero-based indexing). So in the output
above, each MPI rank has access to 1 unique GPU - where MPI 000 has access to GPU 0,
MPI 001 has access to GPU 1, etc., but all MPI ranks show a CUDA runtime GPU ID of 0.
The reason is that each MPI rank only “sees” one GPU and so the CUDA runtime labels
it as “0”, even though it might be global GPU ID 0, 1, 2, or 3. The GPU’s bus ID
is included to definitively show that different GPUs are being used.
Here is a summary of the different GPU IDs reported by the example program:
GPU_IDis the node-level (or global) GPU ID read fromCUDA_VISIBLE_DEVICES. If this environment variable is not set (either by the user or by Slurm), the value ofGPU_IDwill be set toN/A.RT_GPU_IDis the CUDA runtime GPU ID (as reported from, saycudaGetDevice).Bus_IDis the physical bus ID associated with the GPUs. Comparing the bus IDs is meant to definitively show that different GPUs are being used.
So the job step (i.e., srun command) used above gave the desired output. Each
MPI rank spawned 2 OpenMP threads and had access to a unique GPU. The
--gpus-per-task=1 allocated 1 GPU for each MPI rank and the default binding bound
each GPU to the respective task.
Example 2: 8 MPI ranks - each with 2 OpenMP threads and 1 GPU (multi-node)
This example will extend Example 1 to run on 2 nodes. As the output shows, it is a
very straightforward exercise of changing the number of nodes to 2 (-N2) and
the number of MPI ranks to 8 (-n8).
$ export OMP_NUM_THREADS=2
$ srun -N2 -n8 -c2 --gpus-per-task=1 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 000 - OMP 001 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 002 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 001 - OMP 001 - HWT 003 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 002 - OMP 000 - HWT 004 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 002 - OMP 001 - HWT 005 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 003 - OMP 000 - HWT 006 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 003 - OMP 001 - HWT 007 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 004 - OMP 000 - HWT 000 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 004 - OMP 001 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 005 - OMP 000 - HWT 002 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 005 - OMP 001 - HWT 003 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 006 - OMP 000 - HWT 004 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 006 - OMP 001 - HWT 005 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 007 - OMP 000 - HWT 006 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 007 - OMP 001 - HWT 007 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
Example 3: 4 MPI ranks - each with 2 OpenMP threads and 1 *specific* GPU (single-node)
This example will be very similar to Example 1, but instead of using
default binding --gpu-bind=map_gpu will be used to map each MPI rank to a specific GPU.
The map_gpu option takes a comma-separated list of GPU IDs to specify how the MPI ranks are
mapped to GPUs, where the form of the comma-separated list is <gpu_id_for_task_0>, <gpu_id_for_task_1>,....
$ export OMP_NUM_THREADS=2
$ srun -N1 -n4 -c2 --gpus=8 --gpu-bind=map_gpu:0,2,4,6 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 000 - OMP 001 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 032 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 001 - OMP 001 - HWT 033 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 002 - OMP 000 - HWT 002 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 002 - OMP 001 - HWT 003 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 003 - OMP 000 - HWT 034 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 6 - Bus_ID BE
MPI 003 - OMP 001 - HWT 035 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 6 - Bus_ID BE
While this level of control over mapping MPI ranks to GPUs might be useful for some
applications, it is always important to consider the implication of the mapping. For
example, if the order of the GPU IDs in the map_gpu option is reversed, the MPI
ranks and the GPUs they are mapped to would be in different NUMA domains, which
could potentially lead to poorer performance.
$ export OMP_NUM_THREADS=2
$ srun -N1 -n4 -c2 --gpus=8 --gpu-bind=map_gpu:6,4,2,0 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 6 - Bus_ID BE
MPI 000 - OMP 001 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 6 - Bus_ID BE
MPI 001 - OMP 000 - HWT 032 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 001 - OMP 001 - HWT 033 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 002 - OMP 000 - HWT 002 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 002 - OMP 001 - HWT 003 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 003 - OMP 000 - HWT 034 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 003 - OMP 001 - HWT 035 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
Here, notice that MPI 000 now maps to GPU 6, MPI 001 maps to GPU 4, etc., so the MPI ranks are not in the same NUMA domains as the GPUs they are mapped to.
Example 4: 8 MPI ranks - each with 2 OpenMP threads and 1 *specific* GPU (multi-node)
Extending Examples 2 and 3 to run on 2 nodes is also a straightforward exercise by
changing the number of nodes to 2 (-N2) and the number of MPI ranks to 8 (-n8).
$ export OMP_NUM_THREADS=2
$ srun -N2 -n8 -c2 --gpus=16 --gpu-bind=map_gpu:0,2,4,7 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 000 - OMP 001 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 032 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 001 - OMP 001 - HWT 033 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 002 - OMP 000 - HWT 002 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 002 - OMP 001 - HWT 003 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 003 - OMP 000 - HWT 034 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 7 - Bus_ID C7
MPI 003 - OMP 001 - HWT 035 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 7 - Bus_ID C7
MPI 004 - OMP 000 - HWT 000 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 004 - OMP 001 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 005 - OMP 000 - HWT 032 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 005 - OMP 001 - HWT 033 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 006 - OMP 000 - HWT 002 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 006 - OMP 001 - HWT 003 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 4 - Bus_ID 87
MPI 007 - OMP 000 - HWT 034 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 7 - Bus_ID C7
MPI 007 - OMP 001 - HWT 035 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 7 - Bus_ID C7
Mapping multiple MPI ranks to a single GPU
In the following examples, 2 MPI ranks will be mapped to 1 GPU. For the sake of brevity,
OMP_NUM_THREADS will be set to 1, so -c1 will be used unless otherwise specified.
Example 5: 8 MPI ranks - where 2 ranks share a GPU (round-robin, single-node)
This example launches 8 MPI ranks (-n8), each with 1 physical CPU core (-c1)
to launch 1 OpenMP thread (OMP_NUM_THREADS=1) on. The MPI ranks will be assigned
to GPUs in a round-robin fashion so that each of the 4 GPUs on the node are shared
by 2 MPI ranks. To accomplish this GPU mapping, a new srun option will be used:
--ntasks-per-gpuspecifies the number of MPI ranks that will share access to a GPU.--gpu-bind=map_gpuBind tasks to specific GPUs by setting GPU masks on tasks (or ranks) as specified where <list> is <gpu_id_for_task_0>,<gpu_id_for_task_1>,…
$ export OMP_NUM_THREADS=1
$ srun -N1 -n8 -c1 --ntasks-per-gpu=2 --gpu-bind=map_gpu:0,1,2,3 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 002 - OMP 000 - HWT 002 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 003 - OMP 000 - HWT 003 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 004 - OMP 000 - HWT 004 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 005 - OMP 000 - HWT 005 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 006 - OMP 000 - HWT 006 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 007 - OMP 000 - HWT 007 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
Example 6: 16 MPI ranks - where 2 ranks share a GPU (round-robin, multi-node)
This example is an extension of Example 5 to run on 2 nodes.
$ module load craype-network-ucx
$ module load cray-mpich-ucx/8.1.32
$ export OMP_NUM_THREADS=1
$ srun -N2 -n16 -c1 --ntasks-per-gpu=2 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 002 - OMP 000 - HWT 002 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 003 - OMP 000 - HWT 003 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 004 - OMP 000 - HWT 004 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 005 - OMP 000 - HWT 005 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 006 - OMP 000 - HWT 006 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 007 - OMP 000 - HWT 007 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 008 - OMP 000 - HWT 000 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 009 - OMP 000 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 010 - OMP 000 - HWT 002 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 011 - OMP 000 - HWT 003 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 012 - OMP 000 - HWT 004 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 013 - OMP 000 - HWT 005 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 014 - OMP 000 - HWT 006 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 015 - OMP 000 - HWT 007 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
Example 7: 8 MPI ranks - where 2 ranks share a GPU (packed, single-node)
This example launches 8 MPI ranks (-n8), each with 8 physical CPU cores (-c8)
to launch 1 OpenMP thread (OMP_NUM_THREADS=1) on. The MPI ranks will be assigned
to GPUs in a packed fashion so that each of the 4 GPUs on the node are shared by 2
MPI ranks. Packed block distribution appears to be the default for GPU binding; however,
the distribution flag does impact the CPU/Thread binding.
$ export OMP_NUM_THREADS=1
$ srun -N1 -n8 -c8 --ntasks-per-gpu=2 --distribution=block:block ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 008 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 002 - OMP 000 - HWT 016 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 003 - OMP 000 - HWT 024 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 004 - OMP 000 - HWT 032 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 005 - OMP 000 - HWT 040 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 006 - OMP 000 - HWT 048 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 007 - OMP 000 - HWT 056 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
Example 8: 16 MPI ranks - where 2 ranks share a GPU (packed, multi-node)
This example is an extension of Example 7 to use 2 compute nodes. With the appropriate
changes put in place in Example 7, it is a straightforward exercise to change to using
2 nodes (-N2) and 16 MPI ranks (-n16).
$ export OMP_NUM_THREADS=1
$ srun -N2 -n16 -c8 --ntasks-per-gpu=2 --distribution=*:block ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 001 - OMP 000 - HWT 008 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 002 - OMP 000 - HWT 016 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 003 - OMP 000 - HWT 024 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 004 - OMP 000 - HWT 032 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 005 - OMP 000 - HWT 040 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 006 - OMP 000 - HWT 049 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 007 - OMP 000 - HWT 056 - Node defiant-nv01 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 008 - OMP 000 - HWT 001 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 009 - OMP 000 - HWT 008 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 0 - Bus_ID 0A
MPI 010 - OMP 000 - HWT 016 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 011 - OMP 000 - HWT 024 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 1 - Bus_ID 18
MPI 012 - OMP 000 - HWT 034 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 013 - OMP 000 - HWT 040 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 2 - Bus_ID 41
MPI 014 - OMP 000 - HWT 048 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
MPI 015 - OMP 000 - HWT 056 - Node defiant-nv02 - RT_GPU_ID 0 - GPU_ID 3 - Bus_ID 4A
Example 9: Multiple GPUs per MPI rank
This example launches one MPI rank with all available GPUs on the node visible to the rank.
$ export OMP_NUM_THREADS=1
$ srun -N1 -n1 -c1 --gpus-per-task=8 ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0,1,2,3,4,5,6,7 - GPU_ID 0,1,2,3,4,5,6,7 - Bus_ID 0A,18,41,4A,87,90,BE,C7
Example 10: All MPI Tasks to All GPUs
This example show the usage of --gpu-bind=closest which will bind all GPUs to all MPI tasks.
$ export OMP_NUM_THREADS=1
$ srun -N1 -n4 -c1 --gpus-per-task=2 --gpu-bind=closest ./hello_jobstep | sort
MPI 000 - OMP 000 - HWT 000 - Node defiant-nv01 - RT_GPU_ID 0,1,2,3,4,5,6,7 - GPU_ID 0,1,2,3,4,5,6,7 - Bus_ID 0A,18,41,4A,87,90,BE,C7
MPI 001 - OMP 000 - HWT 032 - Node defiant-nv01 - RT_GPU_ID 0,1,2,3,4,5,6,7 - GPU_ID 0,1,2,3,4,5,6,7 - Bus_ID 0A,18,41,4A,87,90,BE,C7
MPI 002 - OMP 000 - HWT 001 - Node defiant-nv01 - RT_GPU_ID 0,1,2,3,4,5,6,7 - GPU_ID 0,1,2,3,4,5,6,7 - Bus_ID 0A,18,41,4A,87,90,BE,C7
MPI 003 - OMP 000 - HWT 033 - Node defiant-nv01 - RT_GPU_ID 0,1,2,3,4,5,6,7 - GPU_ID 0,1,2,3,4,5,6,7 - Bus_ID 0A,18,41,4A,87,90,BE,C7
Note
There are many different ways users might choose to perform these mappings, so users are encouraged to clone the hello_jobstep program and test whether or not processes and threads are running where intended.
NVMe Usage
Each Defiant compute node has [3x] 3.5 TB NVMe devices (SSDs). To use the NVMe, users must
request access during job allocation using the -C nvme option to
sbatch, salloc, or srun. Once the devices have been granted to a job,
users can access them at /mnt/bb/<userid>. Users are responsible for moving data
to/from the NVMe before/after their jobs. Here is a simple example script:
#!/bin/bash
#SBATCH -A <projid>
#SBATCH -J nvme_test
#SBATCH -o %x-%j.out
#SBATCH -t 00:05:00
#SBATCH -p batch-gpu
#SBATCH -N 1
#SBATCH -C nvme
date
# Change directory to user scratch space (/lustre/polis)
cd /lustre/polis/<projid>/scratch/<userid>
echo " "
echo "*****ORIGINAL FILE*****"
cat test.txt
echo "***********************"
# Move file from Lustre to SSD
mv test.txt /mnt/bb/<userid>
# Edit file from compute node
srun -n1 hostname >> /mnt/bb/<userid>/test.txt
# Move file from SSD back to GPFS
mv /mnt/bb/<userid>/test.txt .
echo " "
echo "*****UPDATED FILE******"
cat test.txt
echo "***********************"
And here is the output from the script:
$ cat nvme_test-<jobid>.out
Mon May 17 12:28:18 EDT 2021
*****ORIGINAL FILE*****
This is my file. There are many like it but this one is mine.
***********************
*****UPDATED FILE******
This is my file. There are many like it but this one is mine.
defiant19
***********************
Container Usage
Defiant provides Apptainer v1.2.5 installed for building and running containers. See documentation on how to write Apptainer definition files here . You can also pull images from a registry like Docker Hub, and Apptainer will automatically convert those images to its SIF format.
Note
The container docs will continue to evolve and change as we identify better practices and more user friendly methods for using containers on Defiant to best suit the needs of the users. If something you’re trying no longer works, be sure to come back and check the docs to see if anything has changed.
Build and Run Workflow
As an example, let’s build and run a very simple container image to demonstrate the workflow.
Building a Simple Image
Create a directory called
simplecontaineron home or GPFS andcdinto it.Create a file named
simple.defwith the following contents.Bootstrap: docker From: rockylinux:9 %post dnf install -y wget sudo git gzip gcc openssh hostname
Build the container image with
apptainer build simple.sif simple.def.Apptainer builds the container image in the SIF file format. Unlike Podman, Apptainer gives you a single file for your image that you can later run as your container.
Running a Simple Container in a Batch Job
As a simple example, we will run hostname with the Apptainer container.
Create a file submit.sl with the contents below.
#!/bin/bash #SBATCH -t00:10:00 #SBATCH -A stf007 #SBATCH -N2 #BATCH -P batch #SBATCH -J logs/simple_container_job #SBATCH -o %x_%j.out #SBATCH -e %x_%j.out srun -N2 --tasks-per-node=1 apptainer exec simple.sif hostname
Submit the job with
sbatch submit.sl. This should produce an output that looks like:defiant01 defiant02
Note that if you are running multiple tasks per node, for example with
srun -N1 --tasks-per-node=2 apptainer exec simple.sif hostname, Apptainer is running
an instance of the runtime for each task i.e. the same running container is NOT shared
between multiple tasks running on the same node.
Running an MPI program with an MPI image
For running a program that uses MPI, you will need to build your container image with MPICH. You can find an example in the olcf_containers_examples repository .
Clone the repository
https://github.com/olcf/olcf_containers_examples.Navigate to
olcf_containers_examples/defiant/mpiexample.Run
build.shto build the containers.rocky9mpich412nvidia2411.defbuilds an image based on Nvidia’s CUDA 12.6 container release, and installs MPICH 4.1.2 in it.Submit the submit script with
sbatch submit.sl.You should get output that looks like
<several INFO messages. Can be ignored> ... Hello from rank 1 Hello from rank 0 Hello from rank 2 Hello from rank 3
Getting Help
If you have problems or need helping running on Defiant, please submit a ticket by emailing help@olcf.ornl.gov.
Known Issues
None